You are here
What is EA/TEF?
EA/TEF stands for Esophageal Atresia/Tracheo-Esophageal Fistula (outside of the US, it is called OA/TOF--Oesophageal Atresia/Tracheo-Oesophageal Fistula). The exact numbers are not known, but it is a rare congenital birth defect which affects approximately 1 in 2,500 to 3,500 babies. With EA/TEF, a baby is unable to swallow, and may also have trouble breathing.
Definition
Esophageal atresia (ea) is a birth defect (congenital anomaly) in which the esophagus, which connects the mouth to the stomach, is shortened and closed off (dead ended) at some point along its length. This defect almost always occurs in conjunction with tracheoesophageal fistula (tef), a condition in which the esophagus is improperly attached to the trachea, the "windpipe" that carries air into the lungs. It is believed that these defects occur around the fourth week of pregnancy when the digestive tract is forming. There is no known cause for the defects.
Description
Failure of an unborn child (fetus) to develop properly can result in birth defects. These defects typically involve organs whose function is either incidental or not necessary at all before birth, meaning that the defects will not be detected until the baby is born. The digestive tract is unnecessary for fetal growth, since all nutrition comes from the mother through the placenta and umbilical cord. During fetal development, the esophagus and trachea arise from the same original tissue, forming into two side-by-side passageways, the esophagus leading from the throat to the stomach and digestive tract, and the trachea leading from the larynx to the lungs and respiratory system. Normally, the two tubes form separately (differentiate); however, in the case of ea/tef, they do not differentiate, which results in various malformed configurations. There are five configurations:
- Type A (7.7% of cases): Esophageal atresia in which both segments of the esophagus end in blind pouches. Neither segment is attached to the trachea.
- Type B (0.8%): Esophageal atresia with tracheoesophageal fistula in which the upper segment of the esophagus forms a fistula to the trachea. The lower segment of the esophagus ends in a blind pouch. This condition is very rare.
- Type C (86.5%): Esophageal atresia with tracheoesophageal fistula, in which the upper segment of the esophagus ends in a blind pouch (EA) and the lower segment of the esophagus is attached to the trachea (TEF).
- Type D (0.7%): Esophageal atresia with tracheoesophageal fistula, in which both segments of the esophagus are attached to the trachea. This is the rarest form of EA/TEF.
- Type E (also called Type H) (4.2%): Tracheoesophageal fistula in which there is no esophageal atresia because the esophagus is continuous to the stomach. Fistula is present between the esophagus and the trachea.
Normally, the esophagus moves food from the mouth to the stomach. When the esophagus ends in a pouch instead of emptying into the stomach, food, liquids, and saliva cannot pass through. The combination of ea with tef compromises digestion, nutrition, and respiration (breathing), creating a life-threatening condition that requires immediate medical attention. All babies with ea/tef require surgical repair to correct the condition and allow proper nutrition and swallowing.
Demographics
Esophageal atresia alone or with tracheoesophageal fistula (ea/tef) occurs in approximately one in 4,000 live births.
Causes and symptoms
The cause of esophageal atresia, like that of most birth defects, is unknown. An infant born with ea/tef may initially appear to swallow normally. However, the first signs of ea/tef may be the presence of tiny, white, frothy bubbles of mucus in the infant’s mouth and sometimes in the nose as well. When these bubbles are suctioned away, they reappear. This symptom occurs when the blind pouch begins to fill with mucus and saliva that would normally pass through the esophagus into the stomach. Instead these secretions back up into the mouth and nasal area, causing the baby to drool excessively. Although the infant may swallow normally, a rattling sound may be heard in the chest along with coughing and choking, especially when the infant tries to drink. Some infants, depending on the severity of the defect, may appear blue (cyanosis), a sign of insufficient oxygen in the circulatory system. The infant’s abdomen may be swollen and firm (distended) because the abnormal trachea allows air to build up in the stomach, filling the abdominal space that holds the surrounding organs. Aspiration pneumonia, an infection of the respiratory system caused by inhalation of the contents of the digestive tract, may also develop.
Diagnosis
When a physician suspects esophageal atresia after being presented with the typical symptoms, diagnosis usually begins with gently passing a catheter through the nose and into the esophagus. Esophageal atresia is indicated if the catheter stops at the blind pouch, indicating that it has hit an obstruction. If EA is present, the catheter will typically stop at 4 to 5 inches (10–12 cm) from the nostrils. Barium-enhanced x-ray examination may reveal a dilated esophageal pouch, made larger by the collection of amniotic fluid in the pouch.
During fetal development, the enlarged esophagus may also have pressed on and narrowed the trachea, a condition in the fetus that can contribute to fistula development. Air in the stomach may confirm the presence of fistula; gas in the large intestine rules out intestinal (duodenal) atresia. The physician will also perform a comprehensive physical examination, looking for other congenital anomalies that are known to accompany EA/TEF. Chest x rays may be taken to look for skeletal and cardiac abnormalities. Abdominal x rays may be taken as well to look for intestinal obstruction and abnormalities. An echocardiogram (ECG) may be performed to evaluate heart function and ultrasound of the kidneys performed to evaluate kidney function.
Treatment
Infants with ea, with or without tef, are unlikely to survive without surgery to reconnect the esophagus. The procedure is done as soon as possible; however, prematurity, the presence of other birth defects, or complications of aspiration pneumonia may delay surgery. Once diagnosed, the baby may be fed intravenously until surgery is performed. Mucus and saliva will also be continuously removed via a catheter. Healthy infants who have no complications, such as heart or lung problems or other types of intestinal malformations, can usually have surgery within the first 24 hours of life. Surgery techniques used to treat the five types of ea/tef defects are similar.
Surgery is conducted while the infant is under general anesthesia; a tube is placed through the mouth to continuously suction the esophageal pouch during the procedure. An intravenous line (IV tubing into the veins) is established to allow fluids to be administered as needed during surgery. Usually, the infant is placed on a ventilator, with a tube placed down the airway for at least the length of the surgery.
Typically, the surgeon makes an incision in the right chest wall between the ribs, allowing access to the esophagus and the trachea for repair of one or both as needed. If the gap between the two portions of the esophagus is short, the surgeon may join both ends of the esophagus (anastomosis). If the upper portion of the esophagus is short and a long gap exists between upper and lower portions, reconstructive surgery cannot be performed, and the infant must receive nutrition in some way to allow several months of growth. In this case, a gastrostomy (stomach tube) may be surgically placed directly into the stomach for feeding. In the most typical ea/tef repair, the fistula is first closed off, creating a separate airway. Then the blind esophageal pouch is opened and connected with suturing (stitching) to the other portion of the esophagus, creating a normal “food pipe” directly into the stomach. The esophagus is separated from the trachea if necessary.
If the two ends of the esophagus are too far apart to be reattached, surgury is put off to wait for growth of esophageal tissue, sometimes accompanied by stretching procedures. Surgery to connect the two ends of the esophagus can happen within three to four months of life. Or tissue from the large intestine is used to breech the gap between the two ends.
Nutritional concerns
If an infant is unable to nurse normally before surgery can be performed, nutrition is provided intravenously (parenteral) or directly through a tube into the stomach (gastronomy). After the surgery, infants should be able to swallow normally and resume nursing or feeding. However, a feeding tube (g-tube) may stay in place for months or years, depending on the child’s nutritional needs.
Prognosis
Surgery to correct esophageal atresia is usually successful, with survival rates close to 100 percent in otherwise healthy infants after the condition is corrected. Postoperative complications may include difficulty swallowing, since the esophagus may not contract efficiently, strictures (scar tissue build up) at the surgery site, and gastrointestinal reflux, in which the acidic contents of stomach back up into the lower part of the esophagus, possibly causing ulcers.
Parental concerns
Despite a difficult beginning for infants with esophageal atresia with or without tef, parents can be reassured that the defect can usually be corrected with surgery, allowing normal digestion, nutrition, and breathing to take place in their child. Concerns about complications are well founded, including increased susceptibility to colds and infections, as well as the presence of chronic conditions. Ongoing medical care helps manage these conditions and maintain good health in children who have had ea/tef. Parents can seek advice about strengthening the child’s immune system through appropriate nutrition and supplements.
Below are diagrams of the five different types of EA/TEF.
| TYPE | DESCRIPTION | DIAGRAM |
|---|---|---|
| Type A Esophageal Atresia (7.7%) |
Both segments of the esophagus end in blind pouches. Neither segment of esophagus is attached to the trachea. | 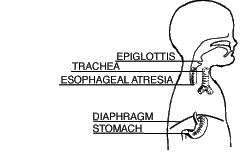 |
| Type B Esophageal Atresia with Upper Tracheoesophageal Fistula (0.8%) |
The upper segment of the esophagus forms a fistula to the trachea (TEF). The lower segment of the esophagus ends in a blind pouch (EA). This is a very rare form of EA/TEF. | 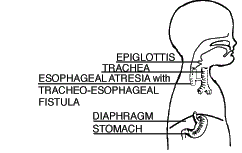 |
| Type C Esophageal Atresia with Lower Tracheoesophageal Fistula (86.5%) |
The upper segment of the esophagus ends in a blind pouch (EA). The lower segment of the esophagus is attached to the trachea (TEF). This is the most common type of EA/TEF. | 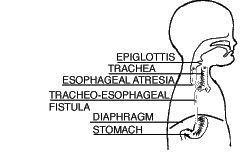 |
| Type D Esophageal Atresia with Upper and Lower Tracheoesophageal Fistula (0.7%) |
Both segments of the esophagus are attached to the trachea. This is the rarest form of EA/TEF. | 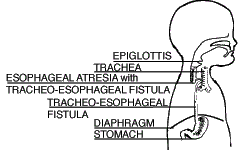 |
| Type E (also called Type H) Tracheoesophageal Fistula (4.2%) |
There is no esophageal atresia as the esophagus is continuous to the stomach. However, fistula is present between the esophagus and the trachea. | 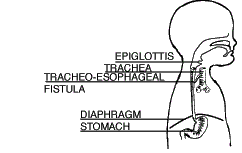 |